News Detail
 来源:本站
来源:本站
 浏览量:19214
浏览量:19214
当前的药物研发是一项高风险的投资,其特点是过程复杂,包括疾病选择、靶标识别、先导化合物发现和优化以及临床前和临床试验。虽然数以百万计的活性化合物已被发现,但是近年来批准新药的数量并没有大幅增加。
在靶点识别和先导化合物发现过程中,会加入激酶谱筛选,进行靶点选择性分析。
根据不同的疾病形成机制,激酶作为药物靶点可以分为以下三类。第一类激酶靶点 不受基因突变或易位的正常调节机制影响,它们具有转化的能力,因此被认为是致癌因素。第二类激酶靶点与另一类非致死突变共同作用抑制蛋白激酶的表达,导致合成致死表型的生成。第三类激酶靶点在肿瘤或其周围组织中表达, 是人类宿体中肿瘤发生和维持的不同阶段所必需的。
成纤维细胞生长因子受体(FGFR)作为受体酪氨酸激酶(RTKs)家族的成员,通过与配体成纤维细胞因子结合并激活下游信号通路参与多种生物学过程的调控,如细胞增殖、迁移、抗凋亡和血管生成等。FGFR基因扩增、错义突变、致癌融合引起的表达及调控异常与多种癌症发生发展有关,FGFR已成为癌症治疗中一个重要的潜在靶点。目前,这些研究大多集中在FGFR1~3上,然而越来越多的证据表明,FGFR4在多种肿瘤的发生和抗肿瘤耐药性的治疗中发挥着重要而独特的作用。FGF19-FGFR4信号通路的异常已被证实是肝癌的致癌因素。
从目前小分子FGFR激酶抑制剂的临床试验情况来看,已有一些小分子抑制剂进入临床研究,其中有8个Lenvatinib、Intedanib、Regorafenib、Ponatinib和Pazopanib,两个为FGFR抑制剂Erdafitinib和Pemigatinib,而其他化合物则处于临床试验、临床前研究和生物活性测试阶段。由于激酶结构域具有相似性,以上多靶点的RTKs抑制剂对FGFR也有一定抑制活性,但同时也出现了较为严重的毒副作用,为了克服脱靶效应,针对FGFR高选择性和高亲和性的小分子抑制剂的研发显得尤为重要。
BLU554是一种高选择性的不可逆共价FGFR4抑制剂,其IC50值为5nmol/L,共价结合到该受体的ATP结合位点。FGF19是FGFR4的配体,在肝癌细胞中高度表达进而促进癌细胞的增殖和分化,BLU-554能够通过抑制FGF19-FGFR4信号通路,诱导肿瘤细胞凋亡,具有好的抗肝癌效果。
验证数据展示:
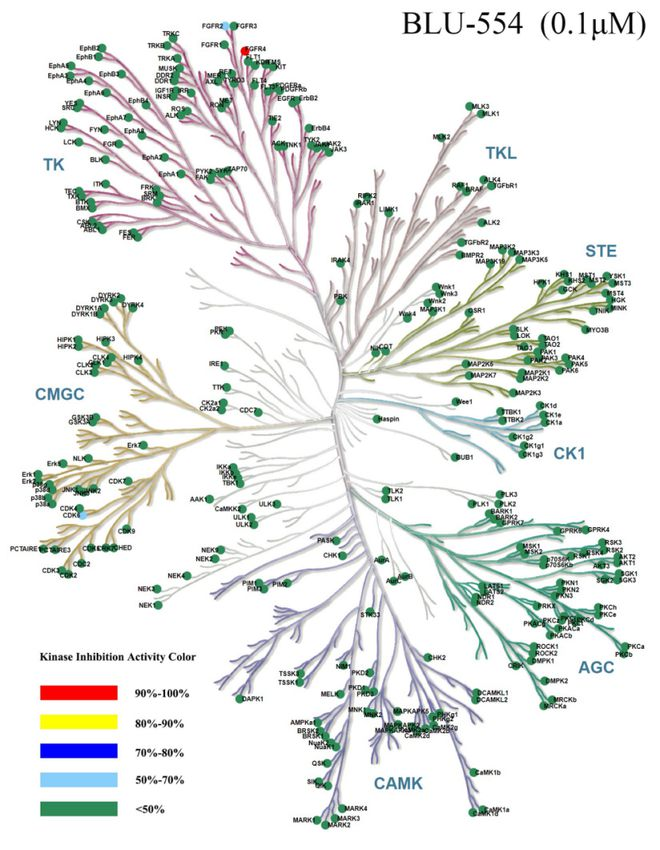
图1 Kinase Panel 416激酶谱树图展示
激酶谱筛选的优势:
l提供激酶谱定制服务
l功能性筛选。
l所有靶点均为人源靶点。
l周周安排检测。
l提供中英文报告,可视化结果展示,全面支持中美IND申报。
l更好的售后服务体验,根据客户投稿要求进行展示图的格式修改。
推出的激酶谱类型:
lCDK激酶谱
lTK酪氨酸激酶谱
l80核心野生型激酶谱
l217野生型激酶谱
l330野生型激酶谱
l416全激酶谱
l定制谱

业务咨询
北京
业务咨询专线:010-6780-9840
联系地址:北京市经济技术开发区科创十三街18号院锋创科技园16号楼
上海
业务咨询专线:010-6780-9840
联系地址:上海市浦东新区蔡伦路780号新技术推广大楼3E5O室
徐州
业务咨询专线:010-6780-9840
联系地址:江苏省徐州市云龙区淮海文博园 二号楼2层
贵州
业务咨询专线:010-6780-9840
联系地址:贵州省贵阳市南明区龙岭路50号 欧美医药产业园一期2号楼