News Detail
 来源:2024-03-19 12:58:29
来源:2024-03-19 12:58:29
 浏览量:21263
浏览量:21263
近年来,ALK(Anaplastic Lymphoma Kinase)基因的改变,包括突变、扩增、融合/重排,在各种恶性肿瘤中的存在已经有了不少报道[1, 2]。其中包括非小细胞肺癌 (NSCLC)、间变性大细胞淋巴瘤 (ALCL)、炎性肌成纤维细胞肿瘤 (IMT)(罕见的中度肿瘤,通常见于儿童,其切除后复发率但转移潜力低)神经母细胞瘤和炎性乳腺癌等。在这些恶性肿瘤里,ALK基因融合/重排突变不太常见,在非小细胞肺癌里ALK基因融合突变的概率是3%,但是其他恶性肿瘤里ALK基因融合/重排突变的概率较低,仅为0.2%。除非小细胞肺癌外,炎性肌成纤维细胞瘤(50% 有 ALK 融合/重排)和间变性大细胞淋巴瘤(50-80%有 ALK 融合/重排)是最常发生 ALK 融合/重排的肿瘤,但基因的融合/重排突变差异很大。在非小细胞肺癌中,大多数肿瘤具有EML4-ALK融合(83.5%),但在其他恶性肿瘤中,这些基因突变/重排占少数(31%)[3-5]。
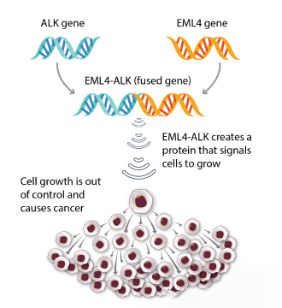
图一:当 ALK 基因与另一个基因(如 EML4)融合(连接)时,就会发生 ALK 融合。融合的基因发出信号,导致细胞生长失控,这意味着 1 个细胞分裂成许多细胞并导致癌症(图片来源于网站https://alk.lungevity.org/)。
间变性淋巴瘤激酶 (ALK) 于 1994 年发现,因为是间变性大细胞淋巴瘤 (ALCL) t(2;5) 染色体易位的融合伴侣,所以ALK 的名称也由此而来[6], 尽管人类 ALK 的酪氨酸激酶结构域与胰岛素受体 (IR) 的酪氨酸激酶结构域高度相似,但还是有所不同。ALK的胞外结构域(ECD)很特别,包含富含甘氨酸的区域(一种低密度脂蛋白) A 类受体 (LDLa) 结构域和 meprin、A-5 蛋白和受体蛋白酪氨酸磷酸酶 mu (MAM) 结构域 (图二)几年后,受ALK基因的调控ALK 受体酪氨酸激酶 (RTK) 被发现,RTK由细胞外配体结合结构域、跨膜结构域和细胞内酪氨酸激酶结构域组成[5-7]当细胞表面的激酶受到刺激时形成磷酸化激酶,可通过信号转导从而影响细胞的增殖和分化,进一步导致癌症的发生。
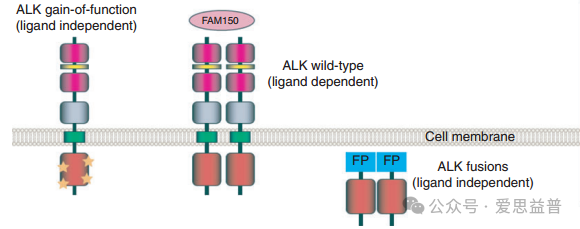
图二:ALK基因的结构图,癌症中的 ALK 信号传导[7]。
ALK 最初被描述为 NPM-ALK 融合体的羧基末端部分。在 NPM-ALK 等融合中,氨基末端融合伴侣(FP,蓝色)与 ALK 的细胞内酪氨酸激酶结构域(红色)融合,导致下游信号传导激活。全长 ALK 受体是一种经典受体酪氨酸激酶,包含氨基末端 ECD 和细胞内酪氨酸激酶结构域(红色),并通过单个跨膜结构域(绿色)连接。ALK ECD 包含两个 MAM 结构域(粉色)、一个 LDLa 结构域(黄色)和一个富含甘氨酸的区域(灰色)。人类 ALK 由小型分泌型 FAM150A (AUGβ) 和 FAM150B (AUGα) 配体激活,这是人类基因组编码的唯一两种被确定含有 FAM150 (AUG) 结构域的蛋白质[6, 7]。全长 ALK 背景下的突变,例如在神经母细胞瘤(金星)中导致激活。ALK 通过许多下游途径发出信号。
近年来,大量的ALK融合蛋白在各种肿瘤中被发现[7]。目前已经有近三十个不同的ALK融合蛋白被报道,其中NSCLC(非小细胞肺癌)中的EML4-ALK和ALCL(间变性大细胞淋巴瘤)中的NPM-ALK得到了广泛的研究。ALK融合蛋白的临床数据主要来自于ALK阳性的NSCLC患者。其中,EML4-ALK是NSCIC中最主要的融合蛋白,代表了大多数的ALK阳性病例。尽管在NSCLC患者中,只有5%会发生ALK的融合,但大量的NSCLC患者使EML4-ALK成为最常见的ALK基因重排,全球每年新发病例约4万例[8-10]。除此之外,KIF5B-ALK, TFG-ALK, KLC1-ALK, PTPN3-ALK, HIP1-ALK, STRN-ALK和TPR-ALK也被报道过。而在ALCL中,有超过50%的病例发生ALK的融合,NPM-ALK是其中最常见的变体,另外还发现了ALO17-ALK, TFG-ALK, MSN-ALK, TPM3-ALK, TPM4-ALK, ATIC-ALK, MYH9-ALK, CLTC-ALK 和TRAF1-ALK等[7, 11]
在各类癌症中,也发现了由于ALK的突变导致的肿瘤发生。已经有超过35种ALK突变被报道。主要是点突变、 ECD的缺失和易位[12, 13][8-9]。这些突变大多位于F1174、F1245、R1275这三个热点残基中的一个[6,11]。ALK的突变会导致神经性母细胞瘤和间变性甲状腺癌等[14]。

图三:ALK阳性所致的不同类别癌症现[7]。
近年来,靶向治疗技术取得了突破性进展,开发出了多种针对ALK的靶向治疗药物。Crizotinib是第一个针对EML4-ALK融合基因的药物,于2011年获准上市[15][13]。研究表明,在晚期EML4-ALK阳性的NSCLC患者中,接受Crizotinib治疗的患者的客观缓解率(ORR;53%)和无进展生存时间(PFS;8.5月)显著高于接受标准铂类化疗的患者[16, 17][14-15]。然而,当Crizotinib作为一线治疗方案使用时,患者通常在治疗后的一年内对其产生不同程度的耐药性[18][16]。因此,第二代EML4-ALK靶向药物,如ceritinib,brigatinib和alectinib,以及第三代靶向药物lorlatinib相继被开发出来,第四代靶向药物repot-rectinib(TPX-0005)一直在进行I/II期临床试验[19][17]。然而,随着靶向治疗的长期使用,不可避免地会出现获得性耐药[20][18]。在NSCLC中,不同的EML4-ALK融合基因变体对靶向药物有不同程度的敏感性[21][19]。因此,对EML4-ALK融合基因进行变异型分析至关重要。研究表明,EML4-ALK获得性耐药机制主要包括以下几个方面:
(1)蛋白激酶结构域的继发性突变。ALK激酶结构域的继发性基因突变导致该激酶与药物结合区的空间构象发生变化,从而增加了该激酶与ATP的结合力,从而影响药物与该激酶的结合,进而导致耐药。第一代靶向药物耐药的患者中,约30%的样本检测到基因突变,导致位于ATP结合区的甘氨酸残基与缬氨酸发生点突变。第二代耐药后的突变率超过50%,导致甘氨酸残基突变为精氨酸[22, 23][20-21]。
(2)激活替代信号通路。当ALK信号通路被靶向药物抑制时,其他促肿瘤的信号通路蛋白被异常激活,,如EGFR和KIT,继续促进肿瘤细胞的增殖[24, 25][22-23]。
(3)上皮-间充质转化。肿瘤上皮细胞向间充质细胞转化增强了肿瘤细胞的侵袭和转移能力[26][24]。在NSCLC EML4-ALK靶向耐药的患者中,间充质标志物vimentin(波形蛋白)的表达增加,上皮标志物E-cadherin(钙黏蛋白E)的表达降低,提示上皮-间充质的转变可能参与了耐药反应[27, 28][25-26]。为了克服肿瘤细胞的耐药性,目前有可能加强EML4-ALK靶向药物与其他抗肿瘤药物的结合,如EGFR抑制剂erlotinib[28] [27],联合使用药物可协同增强抗肿瘤活性和抑制ALK激酶活性[29][28]。
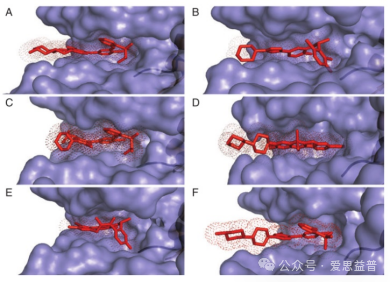
图四:间变性淋巴瘤激酶(ALK)酪氨酸激酶抑制剂(TKIs)在ALK激酶结构域的ATP结合区现[7]。野生型ALK激酶结构域的ATP结合区域的可视化ALK TKIs建模,显示不同的TKI分子之间的接触位点的变化。
A: TAE684 (PDB: 2XB7). B: Crizotinib (PF2341066, PDB: 2XP2). C: Ceritinib (LDK378, PDB: 4MKC). D: Alectinib(CH5424802, PDB: 3AOX). E: Lorlatinib (PF06463922, PDB: 4CLI). F: Brigatinib (AP26113, modelling kindly provided by Tianjau Zhou,ARIAD Pharmaceuticals)
爱思益普Baf3 EML4-ALK细胞编辑株模型介绍
BaF3是一种小鼠白细胞介素-3依赖的pro-B细胞系,BaF3细胞因其对IL-3的依赖性,被广泛用于生物学研究中,尤其是在细胞信号转导和癌症相关研究中。BaF3细胞也被用作重组蛋白的生物学活性测定,例如用于测定某些药物对细胞生长的影响,以及癌症治疗中的药物敏感性试验。是激酶药物发现领域越来越受欢迎的体内外使用的工具细胞。针对ALK靶点,我们在BaF3细胞株上通过编辑成功构建了多种EML4融合的ALK突变细胞模型,用于ALK抑制剂的体内外药物筛选。已有的ALK编辑株及部分验证信息如下:
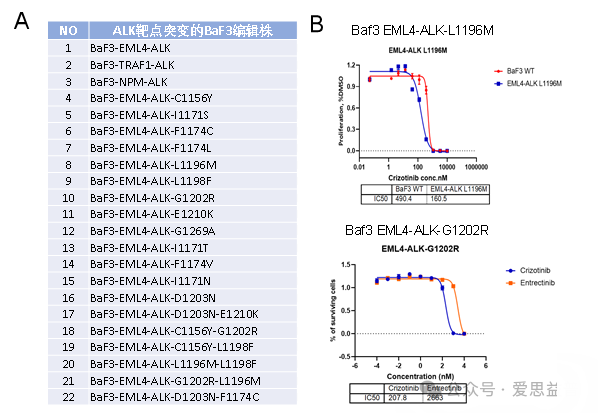
图五:ALK靶点突变的Baf3编辑株体外验证情况。
同时,爱思益普将已构建好Baf3编辑株在小鼠体内构建肿瘤模型,助力于激酶靶点体内药效筛选工作。例如:我们将Baf3 EML4-ALK-G1202R-L1196, Baf3 EML4-ALK-G1202R, Baf3 EML4-ALK-L1196M扩繁后接种到小鼠皮下,观察肿瘤体积和小鼠体重变化,目前已获得成瘤曲线。这些基因编辑的BaF3株能够更好地模拟体内的疾病状态,为药物研发提供更准确的评估。通过这种方法,可以更有效地筛选出候选药物,并为临床治疗提供重要的参考。
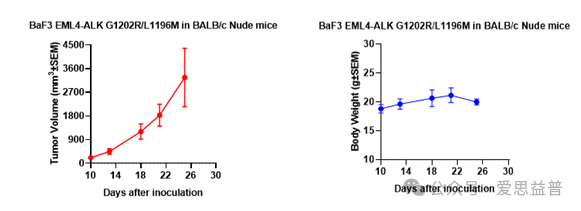
图六:Baf3 EML4-ALK G1202R-L1196成瘤实验。A: 瘤体积变化趋势图。B:小鼠体重变化趋势图
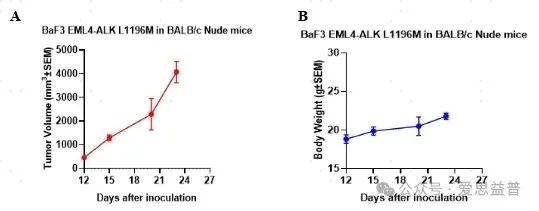
图七:Baf3 EML4-ALK L1196成瘤实验。A: 瘤体积变化趋势图。B:小鼠体重变化趋势图
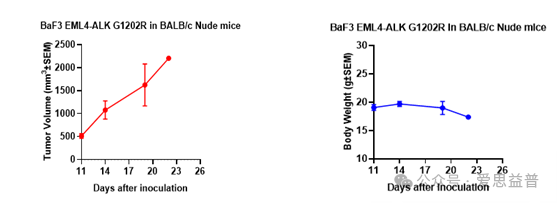
图八:Baf3 EML4-ALK G1202R成瘤实验。A: 瘤体积变化趋势图。B:小鼠体重变化趋势图
1. Soda, M., et al., Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature, 2007. 448(7153): p. 561-6.
2. Davare, M.A., et al., Rare but Recurrent ROS1 Fusions Resulting From Chromosome 6q22 Microdeletions are Targetable Oncogenes in Glioma. Clin Cancer Res, 2018. 24(24): p. 6471-6482.
3. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov, 2017. 7(8): p. 818-831.
4. Ross, J.S., et al., ALK Fusions in a Wide Variety of Tumor Types Respond to Anti-ALK Targeted Therapy. Oncologist, 2017. 22(12): p. 1444-1450.
5. Shreenivas, A., et al., ALK fusions in the pan-cancer setting: another tumor-agnostic target? NPJ Precis Oncol, 2023. 7(1): p. 101.
6. Morris, S.W., et al., Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science, 1994. 263(5151): p. 1281-4.
7. Hallberg, B. and R.H. Palmer, Mechanistic insight into ALK receptor tyrosine kinase in human cancer biology. Nat Rev Cancer, 2013. 13(10): p. 685-700.
8. Chia, P.L., et al., Prevalence and natural history of ALK positive non-small-cell lung cancer and the clinical impact of targeted therapy with ALK inhibitors. Clin Epidemiol, 2014. 6: p. 423-32.
9. Jemal, A., et al., Annual report to the nation on the status of cancer, 1975-2001, with a special feature regarding survival. Cancer, 2004. 101(1): p. 3-27.
10. Bayliss, R., et al., Molecular mechanisms that underpin EML4-ALK driven cancers and their response to targeted drugs. Cell Mol Life Sci, 2016. 73(6): p. 1209-24.
11. Morris, S.W., et al., Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science, 1995. 267(5196): p. 316-7.
12. Okubo, J., et al., Aberrant activation of ALK kinase by a novel truncated form ALK protein in neuroblastoma. Oncogene, 2012. 31(44): p. 4667-76.
13. Fransson, S., et al., Intragenic anaplastic lymphoma kinase (ALK) rearrangements: translocations as a novel mechanism of ALK activation in neuroblastoma tumors. Genes Chromosomes Cancer, 2015. 54(2): p. 99-109.
14. Hallberg, B. and R.H. Palmer, The role of the ALK receptor in cancer biology. Ann Oncol, 2016. 27 Suppl 3: p. iii4-iii15.
15. Fallet, V., et al., [Management of crizotinib, a new individualized treatment]. Bull Cancer, 2012. 99(7-8): p. 787-91.
16. Khan, M., et al., ALK Inhibitors in the Treatment of ALK Positive NSCLC. Front Oncol, 2018. 8: p. 557.
17. Cameron, L.B., et al., Targeted therapy for advanced anaplastic lymphoma kinase (<I>ALK</I>)-rearranged non-small cell lung cancer. Cochrane Database Syst Rev, 2022. 1(1): p. Cd013453.
18. Casaluce, F., et al., Resistance to Crizotinib in Advanced Non-Small Cell Lung Cancer (NSCLC) with ALK Rearrangement: Mechanisms, Treatment Strategies and New Targeted Therapies. Curr Clin Pharmacol, 2016. 11(2): p. 77-87.
19. Lei, Y., et al., EML4-ALK fusion gene in non-small cell lung cancer. Oncol Lett, 2022. 24(2): p. 277.
20. Okada, K., et al., Prediction of ALK mutations mediating ALK-TKIs resistance and drug re-purposing to overcome the resistance. EBioMedicine, 2019. 41: p. 105-119.
21. Heuckmann, J.M., et al., Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin Cancer Res, 2012. 18(17): p. 4682-90.
22. Dagogo-Jack, I. and A.T. Shaw, Crizotinib resistance: implications for therapeutic strategies. Ann Oncol, 2016. 27 Suppl 3(Suppl 3): p. iii42-iii50.
23. Katayama, R., et al., Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med, 2012. 4(120): p. 120ra17.
24. Kunimasa, K., et al., EML4-ALK fusion variant.3 and co-occurrent PIK3CA E542K mutation exhibiting primary resistance to three generations of ALK inhibitors. Cancer Genet, 2021. 256-257: p. 131-135.
25. Kwon, J.H., et al., Afatinib Overcomes Pemetrexed-Acquired Resistance in Non-Small Cell Lung Cancer Cells Harboring an EML4-ALK Rearrangement. Cells, 2019. 8(12).
26. Mittal, V., Epithelial Mesenchymal Transition in Tumor Metastasis. Annu Rev Pathol, 2018. 13: p. 395-412.
27. Shen, J., et al., EML4-ALK G1202R mutation induces EMT and confers resistance to ceritinib in NSCLC cells via activation of STAT3/Slug signaling. Cell Signal, 2022. 92: p. 110264.
28. Guo, F., et al., EML4-ALK induces epithelial-mesenchymal transition consistent with cancer stem cell properties in H1299 non-small cell lung cancer cells. Biochem Biophys Res Commun, 2015. 459(3): p. 398-404.
29. Gelatti, A.C.Z., A. Drilon, and F.C. Santini, Optimizing the sequencing of tyrosine kinase inhibitors (TKIs) in epidermal growth factor receptor (EGFR) mutation-positive non-small cell lung cancer (NSCLC). Lung Cancer, 2019. 137: p. 113-122.

业务咨询
北京
业务咨询专线:010-6780-9840
联系地址:北京市经济技术开发区科创十三街18号院锋创科技园16号楼
上海
业务咨询专线:010-6780-9840
联系地址:上海市浦东新区蔡伦路780号新技术推广大楼3E5O室
徐州
业务咨询专线:010-6780-9840
联系地址:江苏省徐州市云龙区淮海文博园 二号楼2层
贵州
业务咨询专线:010-6780-9840
联系地址:贵州省贵阳市南明区龙岭路50号 欧美医药产业园一期2号楼